
This white paper details the evolution of cell and gene therapies (CGT), explaining the limitations of viral vectors and current CRISPR systems for scalable gene insertion due to issues like DNA toxicity and nuclear delivery. It draws a parallel between CGT’s growth and the antibody market’s history, arguing that innovation in DNA payload design and manufacturability is the key to unlocking the next generation of durable, non-viral therapies.
The first generation of cell and gene therapies was built on viral vectors—and for a time, they were revolutionary. AAV and lentivirus became foundational tools for delivering genetic material into human cells, enabling the approval of life-changing therapies like Zolgensma (for spinal muscular atrophy) and Kymriah (the first CAR-T therapy). But as the field matured, their limitations became harder to ignore. AAVs are size-capped (~4.7 kb), immunogenic, and complex to manufacture. Lentiviruses integrate semi-randomly, raising safety concerns—especially in pediatric populations. Both are expensive and poorly suited for broad, scalable gene insertion.
Over the past decade, the field took a leap forward with the rise of CRISPR and other editing tools. These systems offer precision and programmability—ideal for knocking out genes or making small corrections. The approval of Casgevy (Vertex/CRISPR Therapeutics) for sickle cell disease marked a major milestone. But CRISPR’s potential for inserting whole genes has yet to be realized at scale.
Why? Two key bottlenecks:
Because these problems remain unsolved, some companies have shifted strategy. Generation Bio, for instance, originally aimed to deliver non-viral DNA but ultimately pivoted to RNA-based therapies—which are easier to deliver and avoid DNA-related toxicity, but only operate in the cytoplasm and offer transient expression. These platforms may work for antibody or protein expression, but not for gene replacement or durable editing.
Now, the field is at an inflection point.
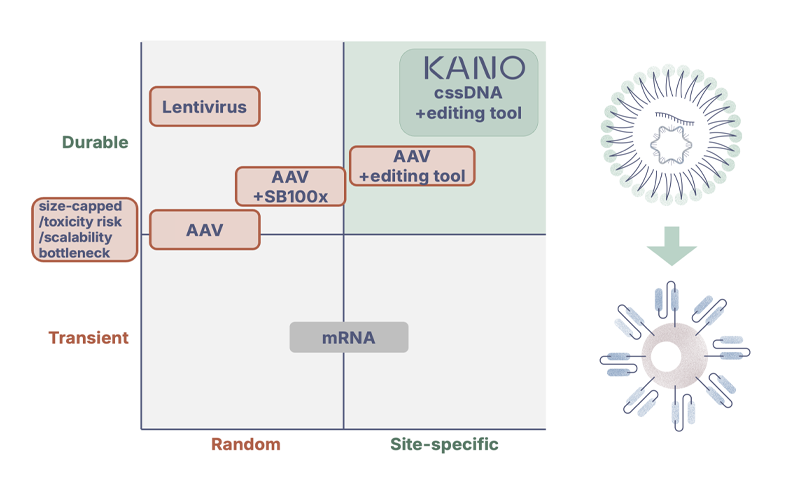
Conceptual landscape of genetic delivery and editing approaches.
The next wave of durable, scalable therapies—like site-specific CAR-Ts, in vivo gene correction for rare diseases, or programmable genomic insertions—requires innovation beyond the editing enzyme. The payload matters. DNA rises from raw material to therapeutic gene product.
Entering the 2nd decade of CGT – should we continue to believe in market growth?
We’re often asked: is Cell and Gene Therapy (CGT) just hype, or are we watching the birth of a lasting therapeutic category? To answer that, it helps to look back — at antibodies. Using sales data from EvaluatePharma, we can overlap the antibody market sales with CGT market sales and projections.
When monoclonal antibodies were first commercialized in the mid-1990s (with ReoPro in 1995), they too faced real skepticism. Yields were low, manufacturing was complex, and early applications were narrow. But as production capabilities improved — from ~0.1 mg/L to 5–10 g/L — the market scaled dramatically. By 2009, 15 years into its journey, the antibody market had grown to nearly $20B annually.
Today, CGT is showing a strikingly similar trajectory. Combined gene and cell therapy sales are projected to reach over $25B by 2030 — just 14 years after the launch of early drugs like Strimvelis (2016) and Luxturna (2017). The resemblance isn’t just in the sales curve — it’s in the evolution of manufacturing, clinical complexity, and regulatory confidence.
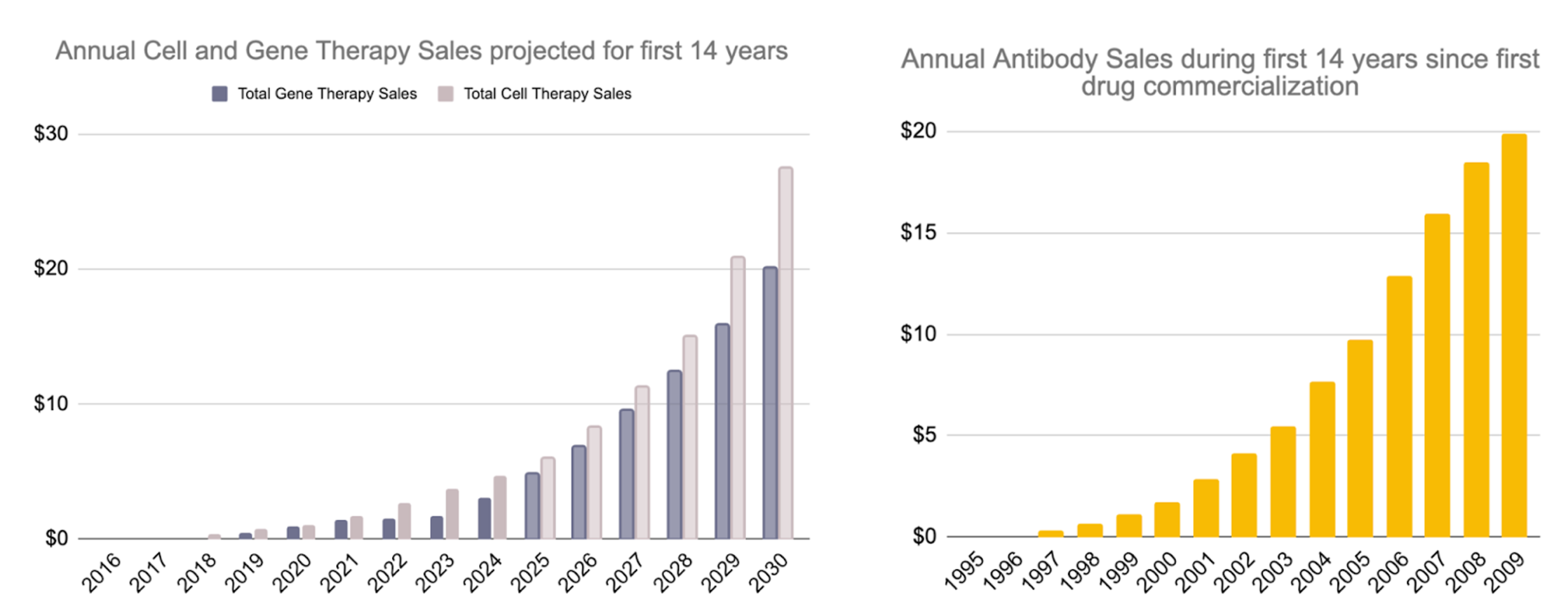
Early commercial trajectories of cell & gene therapies versus antibodies. Based on data from Evaluate Pharma; analysis and visualization by Floris Engelhardt.
At Kano, we see this parallel as both validating and motivating. The CGT field is advancing rapidly, but like antibodies in the early 2000s, it’s being held back by manufacturability — especially when it comes to the DNA payloads required for more complex cell and gene edits. That’s where we come in.
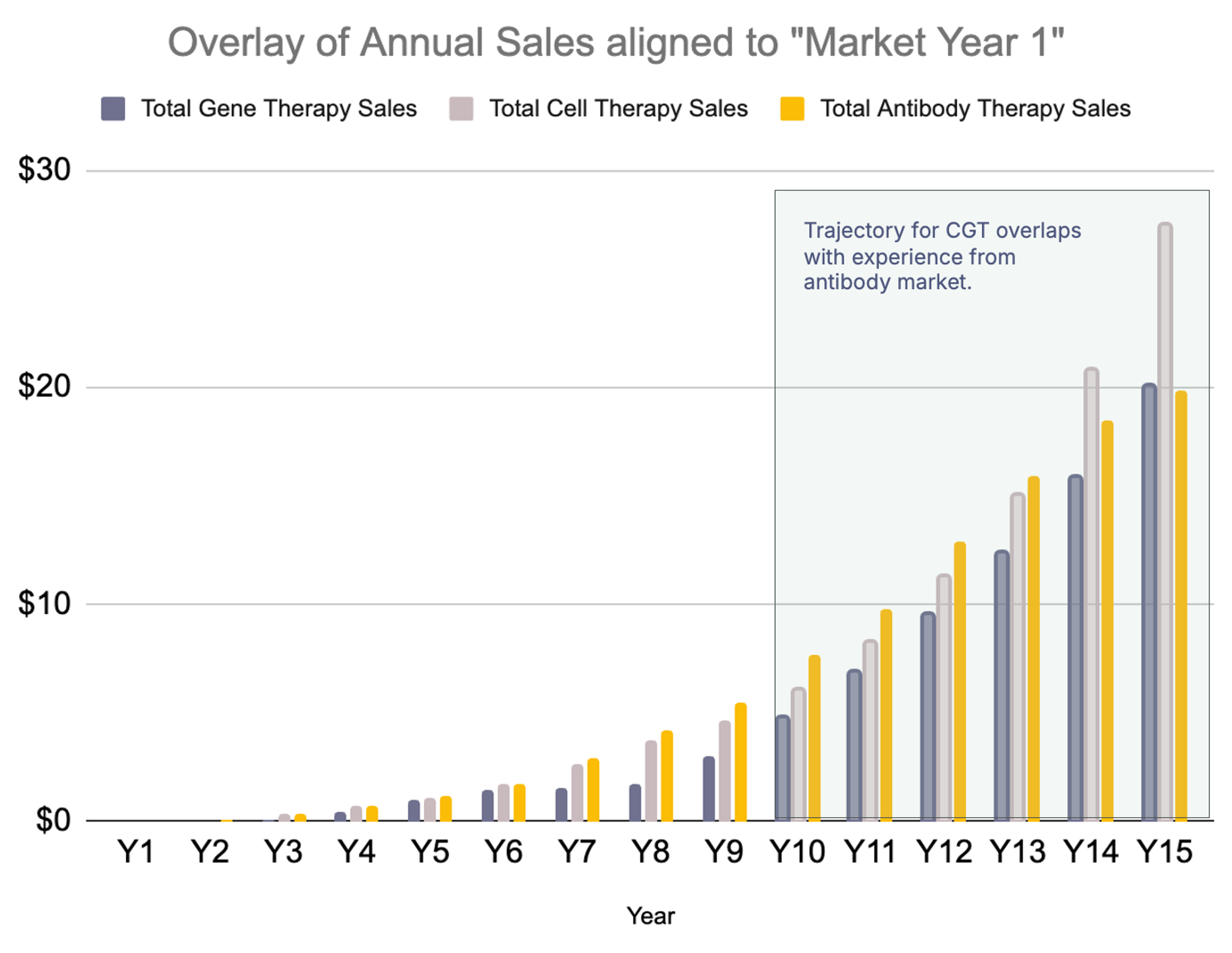
Comparing early market adoption: Overlay of antibody and cell & gene therapy sales aligned to first commercial year. Based on data from Evaluate Pharma; analysis and visualization by Floris Engelhardt.
CGT is entering its second decade. If the antibody playbook holds, the biggest breakthroughs are still ahead — and manufacturability will once again be the catalyst.
Why CAR-T therapies should move from viral to non-viral
Most commercial CAR-T therapies today rely on lentiviral vectors to deliver a chimeric antigen receptor (CAR) transgene into T cells. While this approach has enabled groundbreaking treatments for hematologic malignancies, it has several limitations:
Why multi-target CAR-T and dynamic functionality
Tumor heterogeneity and antigen escape are major causes of relapse in CAR-T-treated patients. Many cancers—especially solid tumors—downregulate or lose the target antigen under therapeutic pressure. Multi-target CAR-Ts and gene circuits address this by:
Together, non-viral CRISPR-based engineering and multi-target CAR design represent the next generation of cell therapies—more precise, adaptable, and better suited to tackle both safety and efficacy challenges across diverse cancers and scalable to other indication profiles.